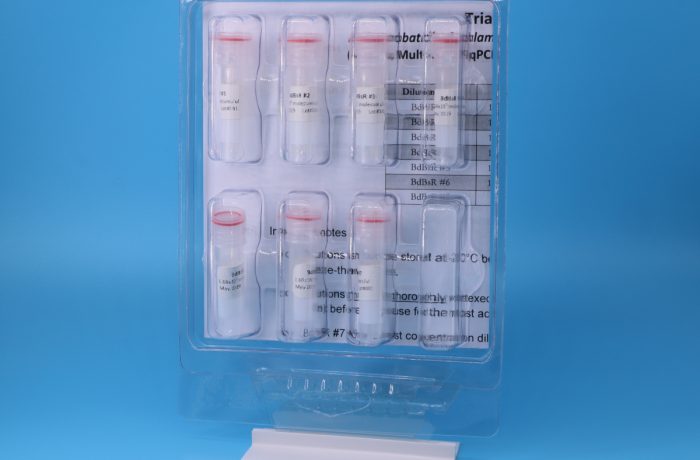
Over time, qPCR assays have steadily superseded PCR assays, both for their ability to provide quantitation of target copy number, as well as decreasing the risk of carry-forward contamination since there is no need to open reaction tubes containing amplified DNA. However, one of the requirements for qPCR assays is a series of (usually) 10-fold serial dilutions of DNA containing the target sequence as positive controls to establish the efficiency and limit of detection of each qPCR run. The importance of qPCR assays for clinically-relevant human pathogens has resulted in the development and commercialization of positive control standards for these assays. This is not the case, however, for most assays for detecting organisms in environmental or wildlife samples. For these assays, individual laboratories running the assay either get positive control DNA from another laboratory or make their own positive in small batches. This results in poorly quantified standards which makes it difficult to compare results between laboratories or even within the same laboratory over time. As part of our work developing and using qPCR assays, we developed plasmid-based positive controls: these are linearized serial dilutions of plasmid DNA containing the target sequence for one (or more) qPCR assays. Plasmid DNA qPCR controls have numerous advantages: (i) they can be produced inexpensively in essentially unlimited quantities, (ii) they can be designed with targets for multiple assays (for example, our Pisces-designed multi-species Gambierdiscus qPCR control), (iii) they are very stable (as part of a NOAA SBIR grant, we incubated a plasmid control kit at 37°C for 13 weeks with no degradation), and (iv) because the assay target sequence in a plasmid is synthetic, it can be made distinguishable from the native genomic sequence, eliminating concerns of false positive samples due to contamination with the controls, unlike native genomic DNA standards. We also developed an improved method for determining the concentration of our plasmid controls: The typical method for quantifying qPCR controls is to measure the amount of DNA and/or calculate the number of molecules from the molecular weight in a concentrated solution, then make serial 10-fold dilutions from this. However, cumulative dilution errors can mean the actual concentration of the final, least concentrated dilution – the most important for determining the limit of detection of the assay – is significantly different than the calculated value. Our improvement was to use similar calculations to get approximate concentrations, then use a “Poisson P0” experiment to measure the exact concentration of the least concentrated dilution. The least concentrated dilution is run in 88 identical qPCR reactions and the percentage that fail (no amplification) is used as the P0 value in a Poisson distribution to calculate the exact average number of target molecules in that dilution. While we use genomic DNA to ground-truth that an assay is working on natural samples and gblocks as quick and easy controls for assays that we run infrequently, our go-to controls for assays we or others run frequently are plasmid-based (we offer a number of these as qPCR control kits).

Applying Molecular Genetics to Solve Challenges in Fisheries, Wildlife and Conservation Science